Wallach's Interpretation of Diagnostic Tests: Pathways to Arriving at a Clinical Diagnosis (589 page)
Authors: Mary A. Williamson Mt(ascp) Phd,L. Michael Snyder Md

An autosomal recessive disorder with abnormal ion transport due to (>1,000) chromosome 7 mutation in the transmembrane conductance regulator (CFTR gene) that controls salt, especially chloride, entry/exit into cells. Incidence of 1:2,500 in non-Hispanic whites in North America with a carrier frequency of 1:20; 1:17,000 in African Americans; marked heterogeneity among patients. For more information, refer to Chapter
10
, Hereditary and Genetic Diseases.
Who Should Be Suspected?
Respiratory symptoms include fatigue, cough, wheezing, recurrent pneumonia or sinus infections, excess sputum and/or shortness of breath.
or
sibling with CF
or
positive neonatal screening AND
or
presence of two
CFTR
genes
or
positive nasal transmembrane potential difference.
Culture:
Special culture techniques should be used in these patients. Before 1 year of age,
S. aureus
is found in 25% and
Pseudomonas aeruginosa
in 20% of respiratory tract cultures; in adults,
P. aeruginosa
grows in 80% and
S. aureus
in 20%.
Haemophilus influenzae
is found in 3.4% of cultures.
Pseudomonas aeruginosa
is found increasingly often after treatment of
Staphylococcus
, and special identification and susceptibility tests should be performed on
P. aeruginosa. Burkholderia cepacia
is becoming more important in older children. Increasing serum antibodies against
P. aeruginosa
can document probable infection when cultures are negative.
Molecular tests
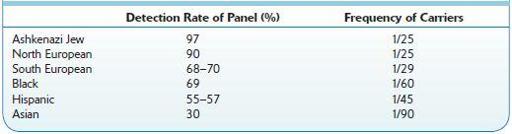
: DNA genotyping (using blood; can use buccal scrapings) to confirm diagnosis based on two mutations is highly specific but not very sensitive and supports diagnosis of cystic fibrosis (CF), but failure to detect gene mutations does not exclude CF because of large number of alleles. A substantial number of patients with CF have unidentified gene mutations. This test should be done when the sweat test is borderline or negative. It can also be used for carrier screening. Identical genotypes can be associated with different degrees of disease severity. The genotype should not be used as sole diagnostic criterion for CF. Prevalence of the 25 most common genes in the panel depends on population group (Table
13-1
). Villus sampling in first trimester or amniocentesis in second or third trimester: >1,000 mutations of
CFTR
gene but the 25 most common account for approximately 90% of carriers. Fifty-two percent are homozygous for ΔF508, and 36% are heterozygous for ΔF508/other CF mutation.
TABLE 13–1. Demographic Groups and Their Risk for Cystic Fibrosis
Core laboratory
: Serum albumin is often decreased (because of hemodilution due to cor pulmonale; may be found before cardiac involvement is clinically apparent). Serum protein electrophoresis shows increasing IgG and IgA levels with progressive pulmonary disease; IgM and IgD levels are not appreciably increased. Serum chloride, sodium, potassium, calcium, and phosphorus levels are normal unless complications occur (e.g., chronic pulmonary disease with accumulation of CO
2
; massive salt loss due to sweating may cause hyponatremia). Urine electrolytes are normal. Submaxillary saliva has slightly increased chloride and sodium but not potassium; considerable overlap with normal results prevents diagnostic use.
Saliva findings
: Submaxillary saliva is more turbid, with increased calcium, total protein, and amylase. These changes are not generally found in parotid saliva.
Other
: Nasal electrical potential difference measurements may be more reliable than sweat tests but are much more complex; mean = −46 mV in affected persons but −19 mV in unaffected persons.
Laboratory changes secondary to complications should also suggest diagnosis of CF:
2
, accumulation of CO
2
, metabolic alkalosis, severe recurrent infection, secondary cor pulmonale, nasal polyps, and pansinusitis; normal sinus x-rays are strong evidence against CF.