Wallach's Interpretation of Diagnostic Tests: Pathways to Arriving at a Clinical Diagnosis (886 page)
Authors: Mary A. Williamson Mt(ascp) Phd,L. Michael Snyder Md

BOOK: Wallach's Interpretation of Diagnostic Tests: Pathways to Arriving at a Clinical Diagnosis
11.09Mb size Format: txt, pdf, ePub
Limitations
The results of a genetic test may be affected by DNA rearrangements, blood transfusion, bone marrow transplantation, or rare sequence variations.
CYSTINE, URINE (CYSTINURIA PANEL)
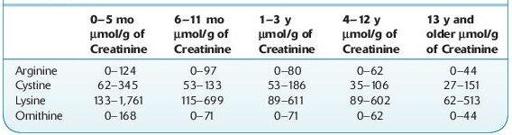
Normal range:
see Table 16.27.
TABLE 16–27. Age-Based Reference Range for Cystine, Arginine, Lysine, and Ornithine
Other books
I Remember You by Harriet Evans
Gypsy Brothers: The Complete Series by Lili St. Germain
Blind Justice by Ethan Cross
Lifetime Guarantee by Gillham, Bill
Once Upon Stilettos (Enchanted Inc #2) by Shanna Swendson
An Unexpected Mother (The Colorado Brides Series Book 4) by Carré White
Ripples on a Pond by Joy Dettman
Hiero Desteen: 01 - Hiero's Journey by Sterling E. Lanier
Clans of the Alphane Moon by Philip K. Dick
An Indelicate Situation (The Weymouth Trilogy) by Church, Lizzie