What Mad Pursuit (8 page)
Authors: Francis Crick

I was still, at this time, a beginning graduate student. By giving my colleagues a very necessary jolt I had deflected their attention in the right direction. In later years few people remembered this or appreciated my contribution except Bernal, who referred to it more than once. Of course in the long run my point of view was bound to emerge. All I did was to help create an atmosphere in which it happened a little sooner. I never wrote up my critique, though my notes for the talk survived for a few years. The main result as far as I was concerned was that Bragg came to regard me as a nuisance who didn’t get on with experiments and talked too much and in too critical a manner. Fortunately he changed his mind later on.
I was, incidentally, not alone on my opinion. In those days most of the other crystallographers believed that protein crystallography was hopeless, or likely to come to fruition only in the next century. In this they were carrying their pessimism too far. I at least had a close acquaintance with the subject and could see one possible method of solving the problem. It is interesting to note the curious mental attitude of scientists working on “hopeless” subjects. Contrary to what one might at first expect, they are all buoyed up by irrepressible optimism. I believe there is a simple explanation for this. Anyone without such optimism simply leaves the field and takes up some other line of work. Only the optimists remain. So one has the curious phenomenon that workers in subjects in which the prize is big but the prospects of success very small always appear very optimistic. And this in spite of the fact that, although plenty appears to be going on, they never seem to get appreciably nearer their goal. Parts of theoretical neurobiology seem to me to have exactly this character.
Fortunately, solving the structure of protein by X-ray diffraction was not as hopeless as it had seemed to some. In 1962 Max Perutz and John Kendrew shared the Nobel Prize for Chemistry for their work on the structures of hemoglobin and myoglobin respectively. Jim Watson, Maurice Wilkins, and I shared the Nobel Prize for Medicine or Physiology in that same year. The citation reads: “. . . for their discoveries concerning the molecular structures of nucleic acid and its significance for information transfer in living material.” Rosalind Franklin, who had done such good work on the X-ray diffraction patterns of DNA fibers, had died in 1958.
The
α
Helix
S
IR LAWRENCE BRAGG was one of those scientists with a boyish enthusiasm for research, which he never lost. He was also a keen gardener. When he moved in 1954 from his large house and garden in West Road, Cambridge, to London, to head the Royal Institution in Albemarle Street, he lived in the official apartment at the top of the building. Missing his garden, he arranged that for one afternoon each week he would hire himself out as a gardener to an unknown lady living in The Boltons, a select inner-London suburb. He respectfully tipped his hat to her and told her his name was Willie. For several months all went well till one day a visitor, glancing out of the window, said to her hostess, “My dear, what
is
Sir Lawrence Bragg doing in your garden?” I can think of few other scientists of his distinction who would do something like this.
Bragg had a great gift for seeing problems in simple terms, realizing that many apparent complications might fall away if the basic underlying pattern could be discovered. It was thus not surprising that in 1950 he wanted to show that at least some stretches of the polypeptide chain in a protein folded up in a simple manner. This was not an entirely new approach. Bill Astbury, the crystallographer, had tried to interpret his X-ray diagrams of keratin (the protein of hair and fingernails) using molecular models with regular repeats. He had found two forms of these fiber diagrams, which he called
α
and
β.
His idea for the
β
structure was not far from the correct answer, but his suggestion for the a structure was completely wide of the mark. This was partly because he was a sloppy model builder and was not meticulous enough about the distances and angles involved and partly because the experimental evidence was misleading in a way that would have been difficult for him to foresee.
It was well known that any chain with identical repeating links that fold so that every link is folded in exactly the same way, and with the same relations with its close neighbors, will form a helix (sometimes incorrectly called a spiral by nonmathematicians). The extreme solutions—a straight line or a circle—are regarded mathematically as degenerate helices.
Bragg’s initial training had been as a physicist, and much of his work on molecular structure had been on inorganic materials such as the silicates. He did not have a detailed familiarity with organic chemistry or the related physical chemistry, though naturally he understood the elements of both these subjects. He decided that a good approach would be to build regular models of the polypeptide backbone, ignoring the complexities of the various side-chains.
A polypeptide chain has as its backbone a regular sequence of atoms, with the repeat . . . CH-CO-NH . . . (where C stands for carbon, H for hydrogen, 0 for oxygen, and N for nitrogen). The actual way that atoms are linked together is shown in appendix A. To each CH is attached a small group of atoms—often called R by chemists, where “R” stands for “Residue.” Here we shall call R the side-chain. We know now that there are just twenty different side-chains commonly found in proteins. For the smallest residue, glycine, R is just a hydrogen atom—hardly a chain at all. The next largest is called alanine and has a methyl group (CH
3
) as its side-chain. The others are of various sizes. Some carry a positive electric charge, some a negative one, and some no charge at all. Most of them are fairly small. The largest two, tryptophan and arginine, have only eighteen atoms in their side-chains. The names of all twenty of them (but not their formulas) are listed in appendix B.
Such a polypeptide chain is built up by joining together little molecules called amino acids. (The details of the chemistry are given in appendix A.) When a protein is synthesized, the relevant amino acids are joined together, head to tail, with the elimination of water, forming a long string called the polypeptide chain. As I have explained, the exact
order
of the amino acids in a particular protein, which is dictated by its gene, determines its character. What we need to know is how each particular polypeptide chain is folded in the three-dimensional structure of the protein and exactly how all the side-chains (some of which are somewhat flexible) are arranged in space, so that we can understand how the protein does its job. Bragg and others wanted to find out, by model building, whether the main polypeptide chain could take up one or more regular folds. There were hints, from Astbury’s
α
and
β
X-ray patterns, that the chain might well do so.
They therefore worked solely with the polypeptide backbone and ignored its side-chains. It may not be obvious why models had to be built at all, since the simple chemical structure of a unit of the backbone was well established. All the bond distances were known and all the bond angles. However, there can be fairly free rotation about bonds called single bonds (but not, by contrast, about double bonds), and the exact configuration of the atoms in space depends on just how these angles of rotation are fixed. This usually depends on interactions between atoms a little distant from one another down the chain and there may be several plausible alternatives, especially if these connections are weak ones.
The reason for this flexibility may not be immediately obvious. An easy way to see this is to use your hand. Place one of your hands so that the fingers are all in one plane, with the thumb exactly at right angles to the index finger. You can still waggle your thumb while preserving this right angle, yet the three-dimensional shape of your hand is changing. (See
figure 5.1.
) This is so even though all the (nearest neighbor) distances are constant—the length of the thumb and of each finger—as are the angles between them. Only the so-called dihedral angle (between the plane of the four fingers and the plane containing your thumb and your index finger) is changing. An example of an “interaction at a little distance” just referred to would be the changing distance between your thumbnail and the nail of your little finger.
In the case of a chemical molecule, there must be interactions of some sort if the molecule has to take up a particular configuration. It was clear that the best way for a polypeptide chain to hold itself together is for it to form hydrogen bonds between certain atoms in its backbone. Hydrogen bonds are weak bonds. The energy is only a small multiple of the thermal energy (at room temperature), and so a single hydrogen bond is easily broken by the constant thermal agitation. This is partly why water is a fluid at normal temperatures and pressures. A hydrogen bond is formed from a donor atom (plus the hydrogen bonded to it) and a recipient. In a polypeptide chain the only strong donor is the NH group, and the only likely recipient the O of the CO group. John Kendrew pointed out that such a hydrogen bond in effect produces a particular ring of atoms. By enumerating all possible rings of this kind one can enumerate all possible structures of this type, each characterized by the NH group bonding to a particular CO group, say one that was three repeats away along the chain. This bonding is repeated over and over again down the length of the chain. The multiple hydrogen bonds thus formed help to stabilize the structure against the battering of thermal motion.
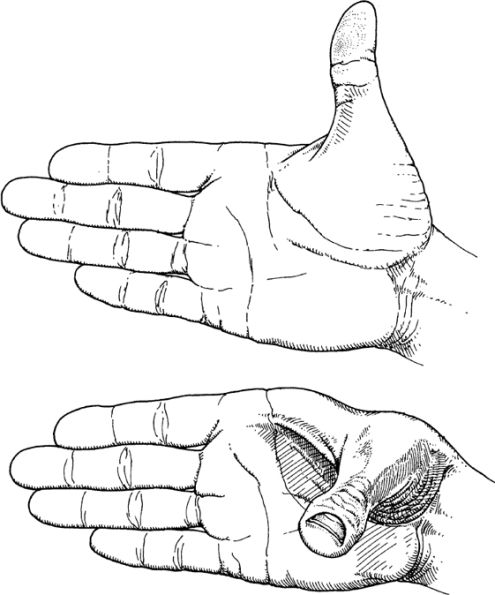
Showing how the thumb can be moved to give a different shape to the hand while preserving all the
direct
angles and distances.
Using special model atoms, made of metal, and links built exactly to scale, Bragg, Kendrew, and Perutz systematically built all possible models, stopping only at folds that were not sufficiently compact. They hoped that one model would prove a much better fit to the X-ray data than all the others. Unfortunately they did not let the models take up their most favorable configurations. Astbury had shown that the α pattern had a strong X-ray spot on the so-called meridian, with a spacing corresponding to a repeat in the fiber direction of 5.1 Å. This implied that an important aspect of the structure repeated after this distance, probably the “pitch”—the distance between successive turns. Because this spot was exactly on the meridian, it suggested that the screw axis (the symmetry element associated with a regular helix) was an integer, though it did not say directly what the integer was. Bragg pointed out that it could be twofold, threefold, fourfold, or even fivefold or higher. As stated earlier, a wallpaper—a two-dimensional repeating pattern—cannot have fivefold symmetry, but there was no reason why a single polypeptide helix should not have a fivefold screw axis. This simply means that if you rotate the helix by 72 degrees (360 degrees divided by 5) and at the same time translate the structure along its axis a certain distance, it will look exactly the same, if you ignore any effects of the ends.
For this reason Bragg, Kendrew, and Perutz built all their models with integer axes. They also built them a little too sloppily. One particular group of atoms, the so-called peptide group, should really be planar—all the six atoms involved should be on or very close to one plane—whereas they allowed rotation about the peptide bond, which made their models too accommodating.
In short, they made one feature (the exact nature of the screw axis) too restrictive and they were too permissive about another—the planarity of the peptide bond. Not surprisingly,
all
their models looked ugly, and they were unable to decide which was best. Reluctantly they published their results in the
Proceedings of the Royal Society
, even though they were inconclusive. It so happened that I was asked to read the proofs of this paper (I believe the proofs were due to arrive when all three authors were away from the lab), but I was too ignorant of the fine points involved to see what was wrong.
Unbeknown to my colleagues, Linus Pauling was also following the same approach. He is now known to the general public mainly because of his championship of vitamin C. At that time he was probably the leading chemist in the world. He had pioneered the application of quantum mechanics to chemistry (explaining in the process, for example, why carbon has a valence of four) and was professor of chemistry at the California Institute of Technology, where he led several very talented groups of research workers. He was especially interested in using organic chemistry to explain important phenomena in biology.
Pauling has described how he first hit on the
α
helix while confined to bed with a cold during his stay in Oxford in 1948 as a visiting professor. His main paper on the
α
helix appeared, with several others of his works, in the
Proceedings of the National Academy of Sciences
in the spring of 1951. Pauling had known that the peptide bond was approximately planar, mainly because he had a more intimate acquaintance with organic physical chemistry than the three Cambridge workers. He had not attempted to make the structure with an integer screw but had let the models fold naturally into any screw they were comfortable with. The a helix turned out to have just 3.6 units per turn. He also noticed a paper by Bamford, Hanby, and Happey, the polymer workers, on the X-ray diffraction of a synthetic polypeptide that fit his model rather well. The fact that his model did not explain the 5.1 Å reflection on the meridian he put to one side. The irony was that Bragg, Kendrew, and Perutz had built, among other models, one that was, in effect, an
α
helix, but they had deformed the poor thing to make it have an exact fourfold axis. This made it look very forced, as indeed it was.
It soon became apparent that Pauling’s α helix was the correct solution. Bragg was quite cast down. He walked slowly up the stairs. (When things went well for Rutherford he would bound upstairs singing “Onward Christian Soldiers.”) “The biggest mistake of my scientific career,” Bragg described it. The fact that it was Linus Pauling who had solved the problem didn’t help, for Bragg had been beaten to the post before by Pauling. Perutz learned that after one of his own seminars a local physical chemist had told him that the peptide group ought to be planar. Perutz had even recorded it on his notes but had done nothing about it. It was not that they had not tried to get good advice, but some of what they had received had been unfortunate. Charles Coulson, a theoretical chemist from Oxford, had told them, in my hearing, that the nitrogen atom might be “pyramidal,” which was a highly misleading piece of information.
Honor was redeemed somewhat when Perutz spotted that the
α
helix should have a strong reflection on the meridian at 1.5 Å, corresponding to the height between successive stages of the helix, and duly found it. Together with two other crystallographers, Vladimir Vand at Glasgow University and Bill Cochran, in the Cavendish, I worked out the general nature of the Fourier Transform of a set of atoms arranged on a regular helix, and Cochran and I showed that it fit rather well the X-ray pattern of a synthetic polypeptide. But in some ways we were rubbing salt into our own wounds..