Power, Sex, Suicide: Mitochondria and the Meaning of Life (27 page)
Read Power, Sex, Suicide: Mitochondria and the Meaning of Life Online
Authors: Nick Lane
Tags: #Science, #General

So there are two good reasons for sustaining poise: keeping respiration as fast as possible, and restricting the leak of reactive free radicals. But maintaining poise is not just a matter of keeping the correct balance of electrons entering the respiratory chains to those leaving at the other end: it also depends on the relative number of carriers within the chains, and this fluctuates because the carriers are continually replaced, like everything else in the body.
Let’s think about this for a moment. What happens if there aren’t enough carriers in the respiratory chains? A shortage of carriers means that the passage of electrons down the respiratory chain slows down, just as too few links in a human bucket chain means there is a slow supply of water getting to the fire. Such a slow transfer of water to the fire equates to a shortage of water: even if the reservoir is full, the house will burn down. Conversely, if there are too many carriers in the middle of the chain, these accumulate electrons faster than they can be passed on down the chain. In the bucket chain analogy, the buckets are being passed faster at the beginning of the chain than at the end—there is a build-up in the middle and everything goes haywire. In both cases, respiration slows down because there is an imbalance in the number of carriers in the respiratory chains, not in the levels of any raw materials. If the concentration of any of these carriers gets out of kilter with the requirements of respiration, respiration slows, and free radicals leak out to cause damage.
Now we are in a position to see why the mitochondria (and chloroplasts too) must retain a contingent of genes of their own. Let’s consider the last carrier of the respiratory chain, cytochrome oxidase, which we met in
Chapter 4
. Imagine that there are 100 mitochondria in a cell. One of these mitochondria does not have enough cytochrome oxidase. As a result, in this mitochondrion, respiration slows down, and electrons back-up in the chains, from where they can
escape to form free radicals. The mitochondrion is inefficient and in danger of damaging itself. To rectify the situation, it needs to make more cytochrome oxidase, so it sends a message to the genes:
Make more cytochrome oxidase!
How would this message operate? The signal might well be the free radicals themselves: a sudden burst of free radicals can alter gene activity through the action of transcription factors that leap into action only when oxidized by free radicals (they are said to be ‘redox-sensitive’). In other words, if there is not enough cytochrome oxidase, electrons back up in the chain and leak out as free radicals. The sudden appearance of free radicals is interpreted by the cell as a signal that there is not enough cytochrome oxidase. It responds accordingly by making some more.
1
Let’s imagine that the genes are in the nucleus. The message arrives, and the nucleus sends orders to make more copies of cytochrome oxidase. It directs the newly minted proteins to the mitochondria using the standard address tag—but this tag can’t discriminate between different mitochondria. As far as the nucleus is concerned, ‘mitochondria’ is a concept and all the mitochondria in the cell share exactly the same address (and it’s quite hard to see how this could be otherwise, given that the mitochondrial population is in a constant state of turnover). So the newly minted cytochrome oxidase is distributed to all 100 mitochondria. The mitochondrion that is short does not get enough. The rest receive too much and so immediately send a message back to the nucleus to say:
Switch off cytochrome oxidase production!
Clearly this situation is untenable. Mitochondria inevitably lose control over respiration, and over-produce free radicals. Cells that lose respiratory control would certainly be selected against. At the very least—and this is a critical point—an inability to control respiration ought to limit the number of mitochondria that a cell could profitably maintain.
Now think what happens the other way round. Imagine that the genes for cytochrome oxidase are retained in the mitochondria. When the signal
Make more cytochrome oxidase!
is sent, it only goes as far as the local gene contingent. These local genes produce more cytochrome oxidase, which is immediately incorporated into the respiratory chains, correcting the imbalance in electron
flow and restoring redox poise. When the message is sent back:
Stop! Switch off cytochrome oxidase production!
it, too, goes only as far as the local gene contingent, and affects only that single mitochondrion. Such a rapid local response could take place in any of the cell’s mitochondria and might in principle operate quite differently in different mitochondria in the same cell at the same time. The cell as a whole retains control over the speed of respiration, and so benefits despite the high costs of maintaining numerous genetic outposts. It would be worse to move the genes to the nucleus.
Professional biochemists or perceptive readers might object at this point. I mentioned in
Part 2
that the respiratory complexes are constructed from a large number of subunits, as many as 45 separate proteins in complex I. Mitochondrial genes encode a handful of these subunits, but the great majority are encoded by nuclear genes. This means that the respiratory complexes are an amalgam encoded by two different genomes. How, then, could a few mitochondrial genes dominate? Surely any construction decisions would need to be shared with the nucleus? Not necessarily. It seems that the respiratory complexes assemble themselves around a few core subunits: once these core proteins have been implanted in the membrane they act as a beacon and a scaffold for the assembly of the rest of the subunits. So if the mitochondrial genes encoded these critical subunits, then they would control the number of new complexes being built. Effectively, the mitochondria make the construction decision, and plant a flag in the membrane; the nuclear components assemble themselves around the flag. Given that the nucleus serves hundreds of mitochondria at once, the overall number of flags in the cell as a whole, at any one time, might remain fairly constant. There would not need to be any change in the overall rate of nuclear transcription to compensate for fluctuations in individual mitochondria, but the effect would be to keep a tight grip on the rate of respiration in all the mitochondria in a cell at once.
If this is true, then Allen’s theory makes some specific predictions about which genes should be retained in the mitochondria. They should encode mostly the core electron-transport proteins in the respiratory chains, such as cytochrome oxidase—those that implant in the membrane like a flag, as if to say ‘
Build here!
’ This is indeed the case (see
Figure 11
). It is also the case for chloroplasts, which as we have seen are in a similar position. Of course, additional genes may also be retained (by chance or for other reasons) but both the mitochondrial and the chloroplast genes of
all
species
always
encode the critical electron-transport proteins, along with the necessary machinery to physically produce the proteins within the mitochondria (such as transfer RNA molecules). When gene loss has progressed to an extreme, it is only—and invariably—this core of respiratory genes that remains. For example, the mitochondria of
Plasmodium
, the cause of malaria, have retained just three proteincoding genes, and as a result they have had to keep all the complex machinery needed to make these proteins in each and every mitochondrion. All three of these genes encode cytochromes—the core electron-transporting proteins of the respiratory chain—exactly as predicted.
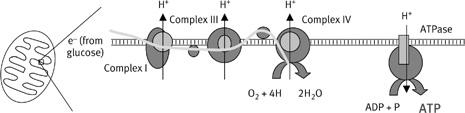
11
Very simplified representation of the respiratory chain, showing the coding of the subunits. Each complex is composed on numerous subunits, about 46 in the case of complex I. Some of these are encoded by mitochondrial genes and some by nuclear genes. John Allen’s hypothesis argues that mitochondrial genes are necessary to control the rate of respiration on a local basis, and for this to work, the subunits encoded by mitochondrial genes should be the core subunits inserted in the membrane. The figure shows that this is broadly true: the subunits encoded by mitochondrial genes (shown in grey) are embedded in the core of the membrane, whereas the subunits encoded by nuclear genes (shown in black) assemble around them. Complex II is not shown here. It does not pump any protons, and does not have any subunits encoded by mitochondrial genes.
The theory makes another prediction, which also seems to be broadly true. This is that any organelles that do not need to conduct electrons will lose their genome. A good example of this is the hydrogenosome of some anaerobic eukaryotes (see
Part 1
,
page 52
). Hydrogenosomes are known to be related to mitochondria, and undoubtedly descend from bacteria. Their function is to carry out fermentations to generate hydrogen gas. They do not conduct electrons, and have no need to maintain redox poise. According to Allen’s theory, they should have no need of a genome—and in virtually all cases they have indeed lost it.
If mitochondria
need
a core of genes to control the speed of respiration, might this explain why bacteria can’t evolve into eukaryotes by natural selection alone? I believe so, although I should emphasize that this is my own speculation (which I have expanded on elsewhere: see Further Reading). Bacteria are about the same size as mitochondria, so clearly a single set of genes can control respiration over a certain area of energetic membranes. Presumably the same is true of bacteria that evolved extensive internal membrane systems, such as
Nitrosomonas
and
Nitrosococcus
. They get by with a single gene set, so presumably that must be enough too. But let’s expand our bacterium; let’s double the area of internal membranes. Now, perhaps, we’re beginning to lose control over some parts of the membrane. If you don’t think so, double the area again. And again. We could double the internal membrane area of
Nitrosomonas
six or seven times before we’re level with the eukaryotes. I doubt we could maintain control over the speed of respiration now. How might we regain control?
One way would be to copy a subset of genes and delegate it to regulate the extra membranes—but how could we choose the right genes? There is no way I can think of that does not involve some kind of foresight (an awareness of which genes to choose), and evolution has none. The only way such delegation might work would be to replicate the entire genome, and then whittle away at one of the two genomes until all the superfluous genes were gone (as actually happened in the mitochondria). But how would we know which genome should lose its genes? Both must be active for genetic control to work. In the meantime, however, we have a bacterium with two active genomes, each under a heavy selective pressure to throw away any excess genes. Either of the two genomes might be expected to lose some genes—but then the two dissimilar genomes would compete with each other, potentially leading to the destruction of the cell (more on this in
Part 6
), and certainly not stabilizing it in the selective battle against other cells.
Such competition between genomes might be stopped if it was possible to demark the sphere of influence of each genome. The eukaryotes solved the sphere-of-influence problem by sealing off the mitochondrial genomes within a double membrane. This is not possible in bacteria, however. If the spare set of genes were sealed off, there would be no way of getting food supplies in and ATP out. In particular, ATP exporters do not exist among bacteria—exporting energy in the form of ATP to their competitors in the outside world would be a suicidal behavioural trait for bacteria. The ATP exporters, along with the family of 150 mitochondrial transport proteins to which they belong, are a eukaryotic invention. We know this because the gene sequences of the ATP exporters are clearly related in plants, animals, and fungi, but there are no similar bacterial genes. This implies that the ATP exporters evolved in the last common ancestor of all the eukaryotes, before the divergence of the major groups, but after the formation of the chimeric ancestral eukaryotic cell.
The eukaryotes had time to evolve such niceties because the relationship between the two partners of the chimera was stable over evolutionary time. The two partners lived in harmony together, and didn’t need anything else—there was ample time and stability for evolutionary change to take place. This stability was only possible because there were other advantages to the association between the collaborating partners. If the hydrogen hypothesis is correct, the
initial advantage was the mutual chemical dependency of two radically different cells, which lasted for long enough for the ATP exporters to evolve. In the case of bacteria evolving simply by natural selection, however, there is no corresponding stability. Simply duplicating a gene set and sealing it off within a membrane could in itself provide no advantages in the interim. Far from it: maintaining extra genes and membranes without any payback is energy sapping, and would no doubt be swiftly dumped by natural selection. Whichever way we look at it, selection pressure is always likely to jettison the burdensome additional genes needed for respiratory control over a wide area of membranes in bacteria. The most stable state is always a small cell that respires across the outer cell membrane. Such a cell will almost invariably be favoured by selection in place of any larger, inefficient, free-radical-generating competitors.